L’esperienza della diagnosi di un tumore cerebrale, dalla comparsa dei sintomi agli approfondimenti radiologici attraverso visite specialistiche è quanto di più coinvolgente e condizionate la vita di una persona. Questa consapevolezza unita a empatia e scienza deve essere ciò che anima lo spirito del Neurochirurgo che si occupa di patologia oncologica e che il paziente sceglie come Specialista a cui affidare le proprie cure, confidare i propri pensieri e con il quale combattere la malattia.
Le modalità di presentazione che in seguito portano alla diagnosi possono essere molteplici e in alcuni casi anche subdole; sono correlate alla sede della massa, all’edema cioè al gonfiore che esibisce il cervello in presenza di una lesione occupante spazio, possono slatentizzarsi in forme di epilessia per un’iperattività elettrica di alcune zone del cervello, possono essere causa di sanguinamenti improvvisi rendendo la diagnosi ancora più difficile.
Per sede si riconoscono i seguenti sintomi e segni:
- Lobo frontale: apatia, abulia, alterazioni comportamentali, irritabilità, cambiamenti del carattere, disinibizione, alterazioni del linguaggio e della produzione verbale, alterazioni nella pianificazione, coordinazione e nell’iniziativa motoria, deficit motori completi o incompleti
- Lobo temporale: alterazioni della sfera emotiva, alterazioni delle funzioni viso-spaziali, alterazioni della comprensione verbale
- Lobo parietale: deficit sensitivi completi o incompleti, alterata percezione del corpo nello spazio circostante
- Lobo occipitale: alterazioni della visione, restringimento del campo visivo, alterata percezione dei colori
Indispensabile, pertanto, è un inquadramento neurochirurgico adeguato fin dal principio e a giudizio del medico, anche eventuali terapie antiedemigene al fine di alleviare i sintomi da lesione occupante spazio: per esempio può essere somministrato il cortisone e in alcuni casi selezionati il mannitolo. Per le forme di epilessia secondaria, cioè causata dalla crescita del tumore stesso, imperativa è l’introduzione di una terapia antiepilettica di copertura.
Tra le indagini di primo livello si menziona la TAC (Tomografia assiale computerizzata): si tratta di una scansione rapida che offre subito una panoramica della situazione ma è un esame di primo livello che prelude ad un altro approfondimento di maggiore dettaglio anatomico ovvero la Risonanza con mezzo di contrasto (RMN).
Completati questi accertamenti e chiarita in prima ipotesi la natura della lesione è indispensabile condividere con il paziente, tramite un ampio colloquio informativo, il percorso terapeutico, la strategia chirurgica e la pianificazione di ulteriori terapie come la chemioterapia e la radioterapia.
La classificazione dei tumori cerebrali universalmente accettata è quella proposta dalla World Health Organization (WHO). Tendenzialmente questa classificazione assegna i tumori del sistema nervoso centrale a categorie ben distinte basate sulla somiglianza istologica delle cellule tumorali alle cellule normali o embrionali. Negli ultimi anni un’ulteriore e più dettagliata classificazione è stata elaborate sulla base delle caratteristiche molecolari esibite dalle cellule tumorali che si è visto avere importanti inferenze prognostiche, ovvero, l’espressione di determinate molecole, enzimi, recettori sulla cellula tumorale condiziona la risposta a trattamenti e la malignità della malattia stessa.
I tumori del sistema nervoso centrale vengono distinti in primitivi cioè derivanti da cellule del sistema nervoso o dei tessuti di sostegno e secondari o metatastici generati cioè dalla migrazione di cellule malate provenienti da altri distretti del corpo.
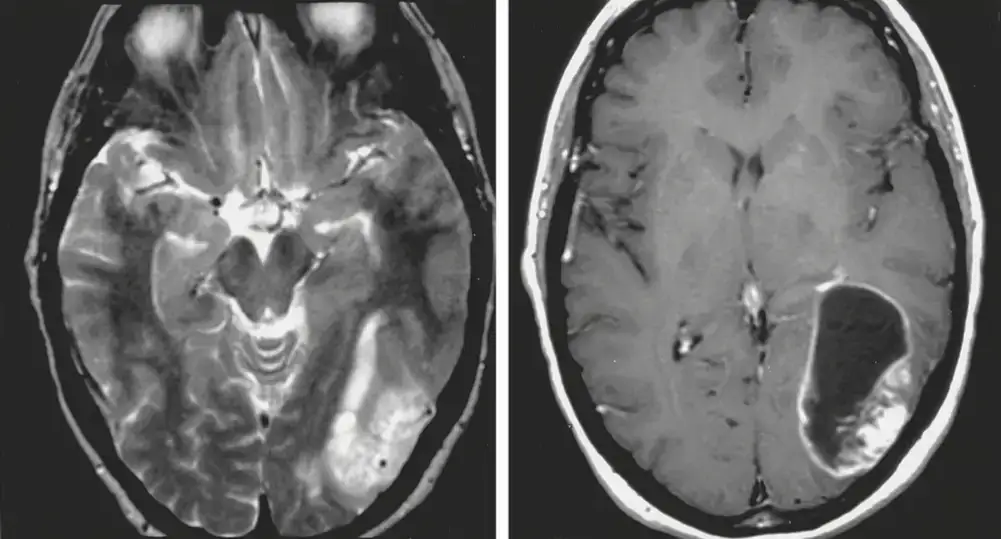
Glioma di alto grado temporale posteriore sinistro. A sinistra sequenza assiale T2 RMN. A destra sequenza RMN assiale T1 con mezzo di contrasto.
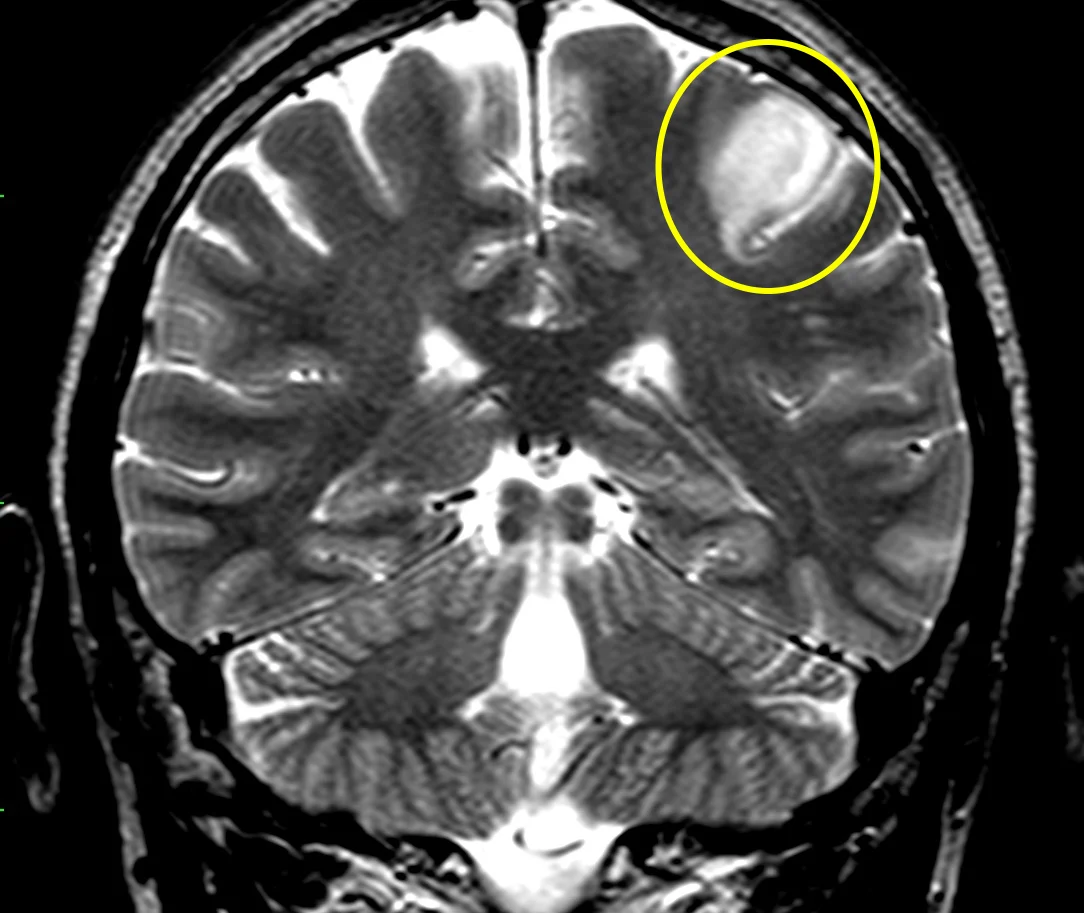
Glioma di basso grado: sequenza RMN T2, lesione cortico sottocorticale sinistra
I tumori primitivi comprendono la grande categoria dei tumori neuroepiteliali all’interno dei quali sono raggruppati:
- Astrocitomi o gliomi (astrocitoma pilocitico, astrocitoma diffuso, astrocitoma anaplastico, glioblastoma) derivanti dagli astrociti ovvero cellule di supporto
- Oligodendrogliomi derivanti dagli oligondendrociti ovvero cellule di supporto
- Ependimomi (ependimoma, ependimoma anaplastico, subependimoma) derivanti dalle cellule ependimali ovvero l’epitelio che avvolge le cavità ventricolari
- Tumori del plesso corioideo (papilloma, carcinoma del plesso corioideo) derivanti dall’epitelio dei plessi corioidei responsabili del turn over del liquido cerebrospinale all’interno delle cavità ventricolari e negli spazi liquorali
- Tumori neuronali o misti glioneuronali (gangliocitoma, ganglioglioma, DNET, neurocitoma) a derivazione puramente neurale o composizione eterogena
- Tumori della regione pineale (pinealomi, pinealoblastomi, tumori a cellule germinali) derivanti dal tessuto della ghiandola pineale oppure da cellule germinali qui rappresentate
- Tumori di derivazione embrionale: medulloblastoma e tumore neuroectodermico primitivo o PNET
Tra tutti i tumori neuroepiteliali spiccano gli astrocitomi o gliomi.
Esistono diversi tipi istologici o sottotipi di gliomi: in realtà l’innovazione nel campo della biologia molecolare ha aperto nuove prospettive anche sulle caratteristiche molecolari di questi tumori superando i retaggi istologici. La nuova classificazione infatti li identifica con nomenclature specifiche che, come detto in precedenza, permettono inferenze prognostiche.
Queste sigle in base all’espressione molecolare sono: IDH, MGMT, codelezione 1p19 q, istone K27M, ecc.
Vedi il PDF “Gliomi Subclassifications 2017”
Vedi il PDF “Gliomi WHO Classification 2021”
Essi possono manifestarsi come una lesione relativamente focale (e in questi casi il comportamento è generalmente benigno: astrocitoma pilocitico) oppure diffusamente infiltranti con una tendenza intrinseca alla degenerazione maligna (astrocitoma diffuso, astrocitoma anaplastico, glioblastoma).
Anche l’età del paziente correla con il tipo di lesione gliale e la sua localizzazione: negli adulti i gliomi tendono ad essere maligni con coinvolgimento emisferico, al contrario l’astrocitoma pilocitico è una neoplasia dell’infanzia e dell’adolescenza molto comune a livello del cervelletto e del terzo ventricolo e meno rappresentata in sede emisferica.
I tumori gliali, specie se altamente infiltranti, hanno una tendenza intrinseca alla disseminazione attraverso le aree compatte di sostanza bianca come il corpo calloso (un insieme di fasci a comunicazione tra i due emisferi) e anche alla disseminazione liquorale (attraverso il liquido cerebrospinale), ependimale (attraverso le cellule ependimali che tappezzano i ventricoli cerebrali), piale (attraverso la sottilissima membrana che aderisce al cervello) e attraverso gli spazi perivascolari.
È evidente che, sulla base di queste considerazioni, fondamentale è il corretto inquadramento clinico, la tempestività della diagnosi e la pianificazione chirurgica: la radicalità dell’asportazione chirurgia infatti condiziona la prognosi. Un’exeresi radicale non solo è curativa per alcuni istotipi (astrocitoma pilocitico) ma nelle forme più infiltranti e invasive (astrocitoma diffuso, astrocitoma anaplastico, glioblastoma) è il presupposto per l’azione e l’efficacia di terapie successive come la radioterapia e la chemioterapia, aumentando la sopravvivenza e contenendo il rischio di recidiva di malattia.
Meningioma
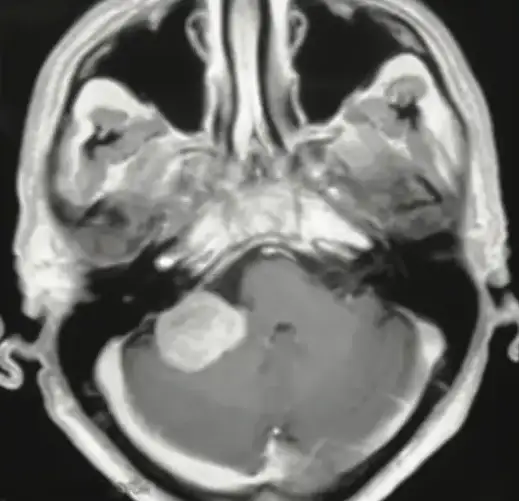
Menigioma della faccia posteriore della rocca: Sequenza RMN assiale T1 con mezzo di contrasto di meningioma della faccia posteriore della rocca a destra.
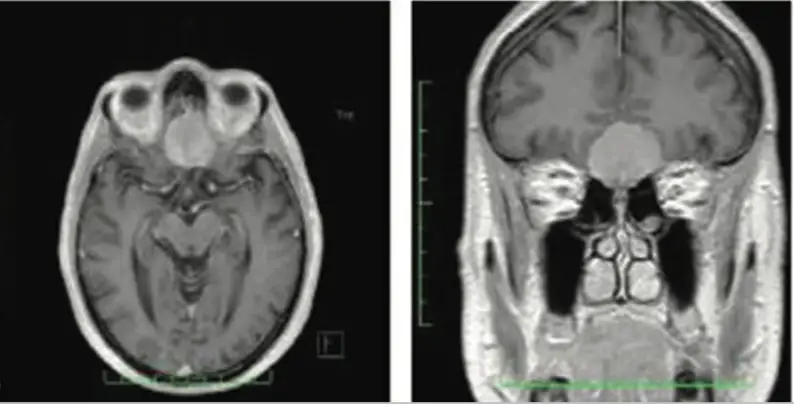
A sinistra sequenza RMN assiale T1 con mezzo di contrasto di meningioma a partenza dalle docce olfattorie. A destra sequenza RMN coronale T1 con mezzo di contrasto di meningioma delle docce olfattorie.
Appartenenti sempre alla categoria dei tumori primitivi vi sono i tumori delle meningi: si tratta di tumori tendenzialmente benigni che originano dalle cellule meningoteliali della dura madre. Chiamati comunemente meningiomi, prediligono le sedi in cui vi è un contatto diretto con i foglietti meningei (dura madre, tentorio) da cui originano ma in alcuni casi possono originare anche dall’aracnoide (una plicatura meningea più profonda e adesa al cervello) e quindi avere anche altre localizzazioni. Per queste lesioni la chirurgia, quando indicata, è curativa fatta eccezione per alcuni sottotipi che possono esibire maggiore tendenza alla recidiva e quindi necessitare di controlli post-operatori ravvicinati ed eventuali terapie complementari.
Adenoma ipofisario
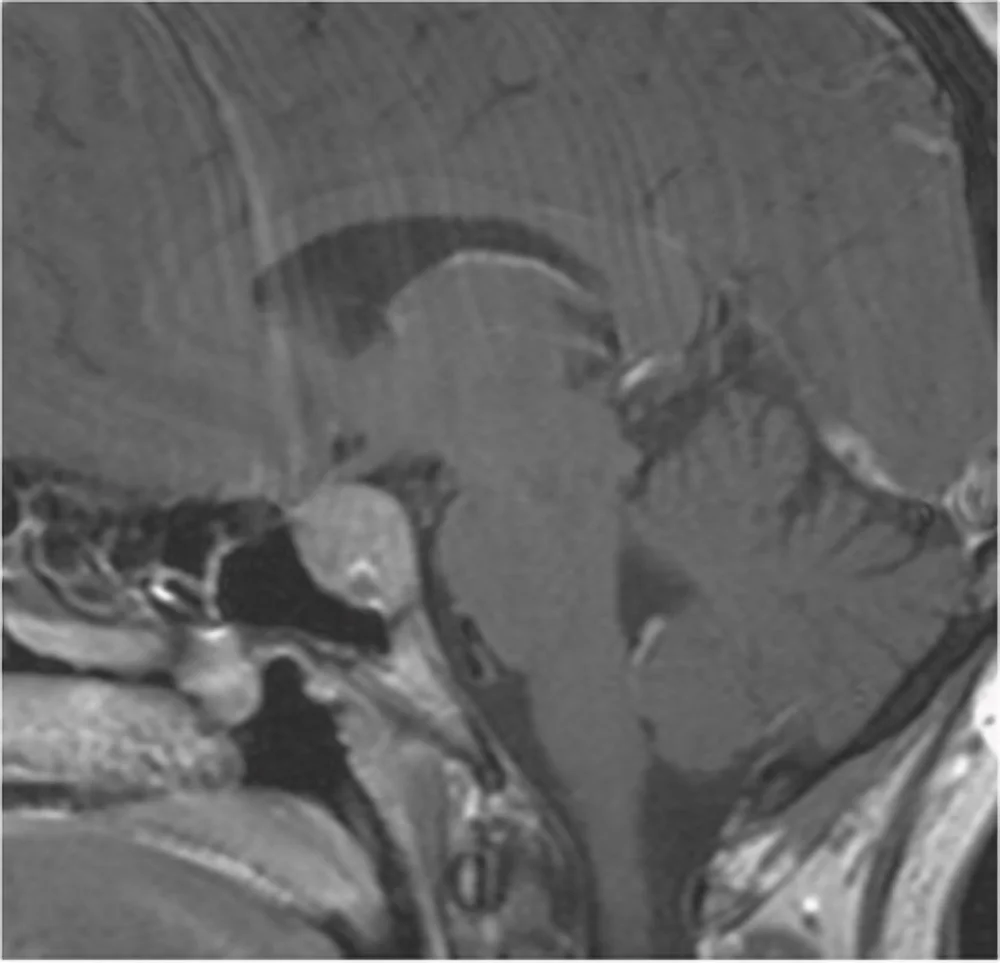
Adenoma ipofisario: sequenza sagittale RMN T1 con mezzo di contrasto, si apprezza una lesione sellare aggettante nel seno sfenoidale.
Adenoma ipofisario: sequenza coronale RMN T1 con mezzo di contrasto, la lesione sellare appare a ridosso del tratto cavernoso delle due arterie carotidi interne.
Tra i tumori primitivi vi sono anche i tumori della regione sellare o adenomi ipofisari. Si tratta di lesioni benigne che derivano dalle cellule della ghiandola ipofisaria.
Il paziente in questi casi manifesta comunemente un disturbo visivo noto come emianopsia bitemporale. Si tratta di un restringimento o ablazione completa della visione nei settori temporali di entrambi gli occhi a causa del rapporto di vicinanza che queste lesioni contraggono con il chiasma ottico, una struttura anatomica in cui le fibre di ciascun nervo ottico in parte si incrociano e in parte decussano.
Normalmente questi tumori vengono distinti in secernenti e non secernenti in base al pattern di secrezione di alcuni ormoni (Prolattina PRL, Ormone Somatotropo GH, Ormone Adrenocorticotropo ACTH, Ormone Tireotropo TSH, Ormone Follicolo Stimolante FSH). Pertanto, possono manifestarsi con disturbi endocrinologici legati all’iperproduzione ormonale. Alternativamente sono masse che non determinano alcuno squilibrio endocrinologico poiché derivanti da cellule non secernenti.
Comunemente si parla di microadenomi < 10 mm e macroadenomi > 10 mm.
Considerata la sede, fondamentale è l’inquadramento e la diagnosi differenziale con lesioni di tipo infiammatorio, ipofisiti anche su base autoimmunitaria, iperplasia ipofisaria molto comune nelle donne in gravidanza o in fase post partum, apoplessia ipofisaria ovvero una nacrosi massiva con saguinamento della ghiandola ipofisaria (evento acuto, improvviso, associato a insufficienza secretiva della ghiandola e pertanto meritevole di trattamento in urgenza), lesioni cistiche derivanti da residui embrionali (cisti della tasca di Rathke) e craniofaringiomi (tumori di derivazione da residui epiteliali della tasca di Rathke con cisti nel contesto e aventi un pattern bimodale di presentazione: le forma adamantinomatose in età pediatrica e anziana, le forme papillari più tipiche dell’età adulta).
Neurinoma
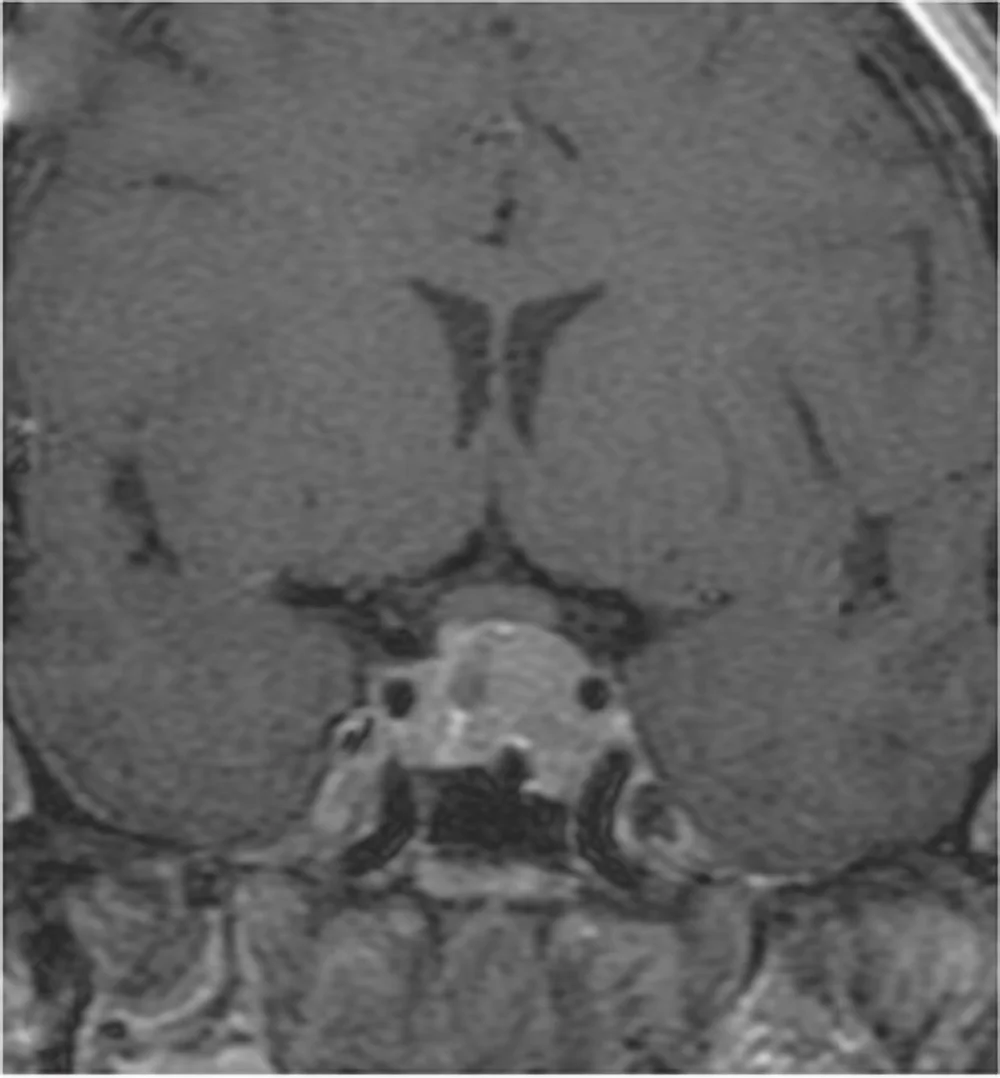
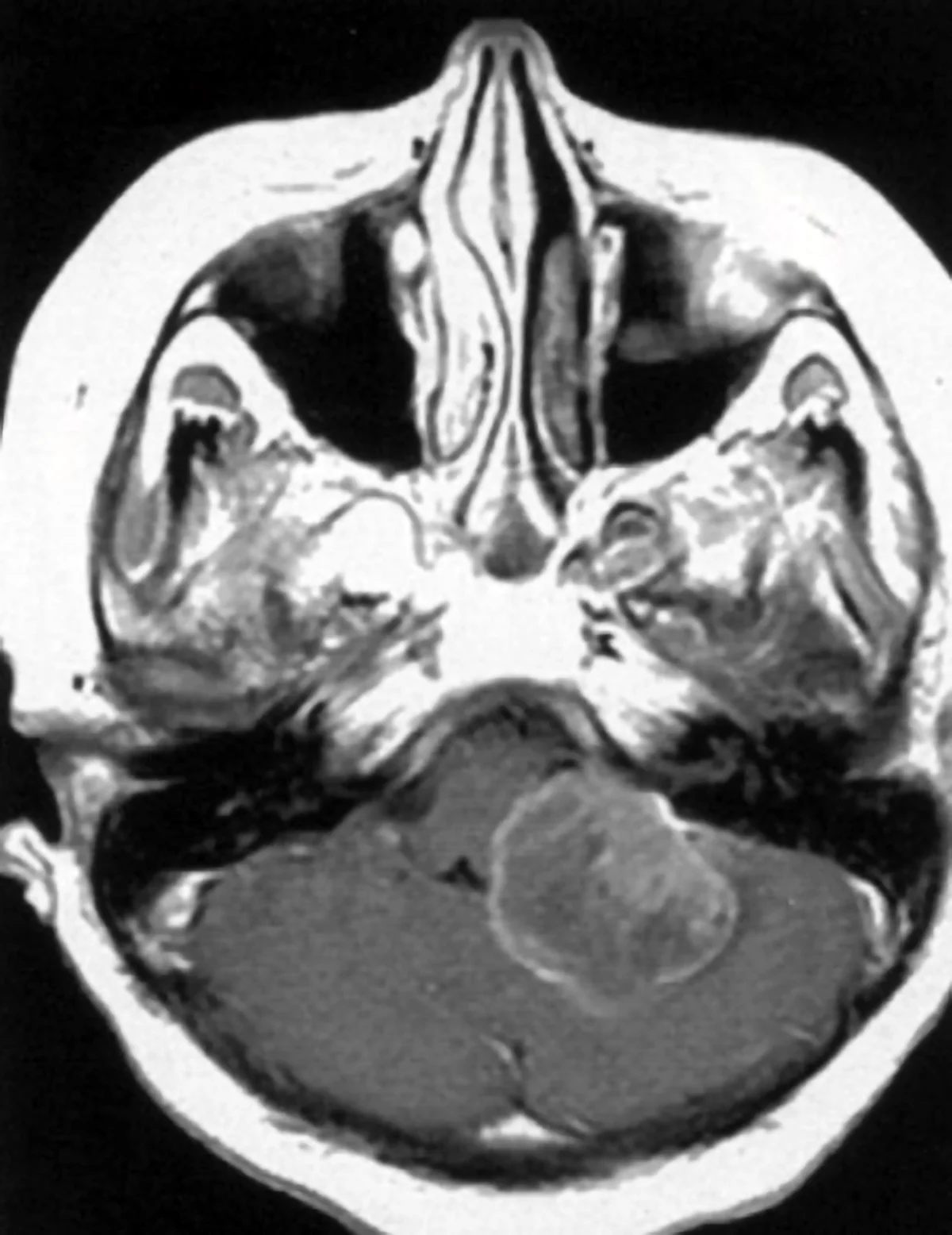
Neurinoma gigante dell’angolo ponto cerebellare destro: Sequenza RMN cisternale assiale.
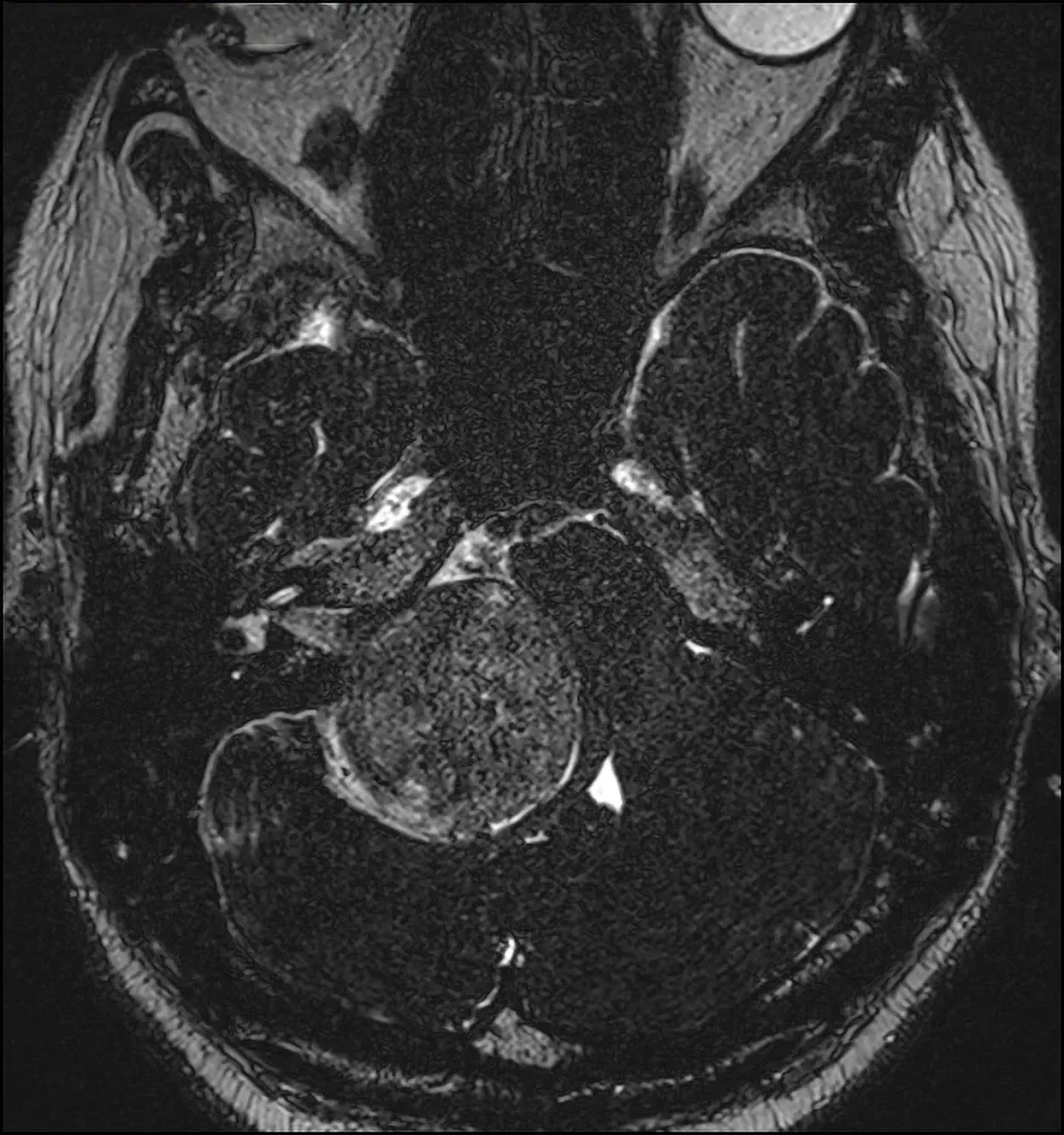
Neurinoma gigante dell’angolo ponto cerebellare destro: sequenza RMN T1 con mezzo di contrasto, caratteristica forma a “cono gelato”con estensione intra-extrameatale.
Tra i tumori primitivi si citano anche i tumori delle guaine nervose o neurinomi o schwannomi.
Si tratta di tumori benigni ben capsulati che originano da cellule di Schwann responsabili del rivestimento mielinico dei nervi cranici. Possono manifestarsi sporadicamente o come parte di sindromi neurocutanee come la Neurofibromatosi di tipo 1 e 2. Trattandosi di tumori della guaina nervosa, tutti i nervi cranici possono essere interessati, ma le forme più frequenti sono neurinomi del III nervo cranico (oculomotore), neurinomi del V nervo cranico (trigemino), neurinomi dell’VII-VIII nervo cranico (pacchetto acustico facciale).
Pur essendo tumori benigni il rapporto con queste strutture nervose può comportare la manifestazione con deficit neurologici: ptosi oculare (III), nevralgia facciale (V), denervazione dei muscoli masticatori e parestesie facciali (V), acufeni, vertigini, sordità, paralisi o asimmetrie facciali (pacchetto acustico – facciale, VII-VIII).
L’opzione chirurgica pertanto, pur essendo risolutiva, richiede una tecnica neurochirurgica di comprovata esperienza che attraverso un’accurata dissezione e identificazione dei piani anatomici, sempre con l’ausilio del monitoraggio intraoperatorio, consenta un’asportazione totale o subtotale garantendo l’integrità della funzione nervosa. Infine, in casi selezionati, si possono proporre altri tipi di terapie ablative come la radiochirurgia stereotassica spiegando al paziente i potenziali rischi e benefici della stessa.
Linfoma
Per quanto concerne il capitolo dei linfomi, il linfoma primitivo è considerato un linfoma maligno a partenza primitiva dal sistema nervoso centrale e pertanto deve essere distinto da malattie linfomatose sistemiche con coinvolgimento secondario dell’encefalo. Il linfoma primitivo del sistema nervoso centrale (PCNSL Primary Central Nervous System Lymphoma) si presenta sia in pazienti immunocompetenti che immunodepressi, può manifestarsi come lesioni singole o multiple spesso localizzate negli emisferi cerebrali; nel 95% dei casi si tratta di linfomi a cellule B. La diagnosi precoce è fondamentale: la chirurgia riveste un ruolo margine spesso utile come conferma diagnostica e istologica. Come per i linfomi sistemici le opzioni terapeutiche comprendono chemioterapia, radioterapia e terapia cortisonica. La percentuale di risposta a queste terapie è molto elevata (70-80%) ma concreto è anche il rischio di recidiva di malattia.
Metastasi
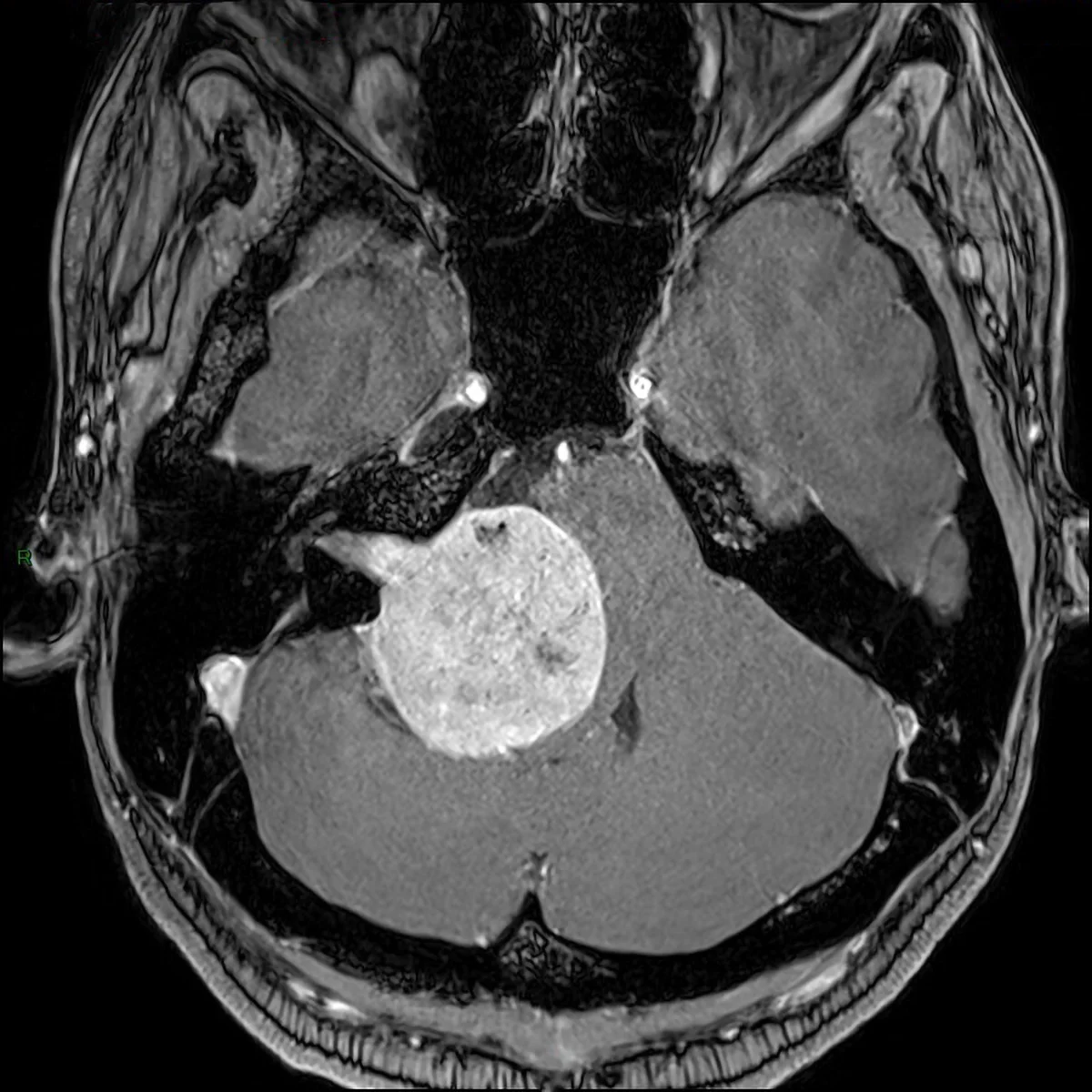
Metastasi in fossa cranica posteriore: sequenza RMN assiale T1 con mezzo di contrasto, lesione a livello del peduncolo cerebellare medio sinistro con compressione del tronco encefalico e del IV ventricolo (metastasi da carcinoma polmonare).
Le metastasi sono tumori secondari che si sviluppano da tumori primitivi verso un’altra sede. Le metastasi al sistema nervoso centrale diffondono prevalentemente per via ematica. Altre vie di disseminazione sono le disseminazioni per contiguità che possono essere perivascolari oppure perineurali, ovvero vie di minore resistenza dove l’osso è più sottile o assente come forami naturali o fessure craniche attraverso cui decorrono vasi e nervi. La formazione di una metastasi cerebrale, tuttavia, è un fenomeno biologicamente complesso e geneticamente mediato in quanto comporta che, parimenti alla colonizzazione dell’habitat, vi sia la creazione di un microambiente in grado di favorire la crescita del tumore: attivazione di proto-oncogeni (geni che favoriscono il tumore), disattivazione di geni oncosoppressori (geni che combattono il tumore), meccanismi pro-angiogenetici che supportano l’attivo metabolismo tumorale.
Nel 10% dei casi il tumore primitivo risulta essere ignoto alla prima diagnosi di metastasi cerebrale e nel 10% dei casi il cervello è l’unico organo colpito. Il tessuto cerebrale nell’80% dei casi è la sede più interessata in particolare gli emisferi cerebrali, a seguire l’osso cranico e la dura madre nel 15%. Le infiltrazioni diffuse leptomeningee (aracnoide e pia madre) fino a quadri di carcinomatosi meningea sono molto rare (5%).
Nel 50% dei casi la metastasi cerebrale è unica, nel 30% sono presenti più di tre lesioni, nel 5% più di cinque lesioni.
L’origine più frequente è il tumore al seno, al polmone, il tumore renale, il tumore del colon e il melanoma.
La chirurgia delle metastasi trova un razionale solo nel caso di metastasi solitaria o metastasi adiacenti fermo restando che l’intervento chirurgico non agisce sulla malattia sistemica che ha portato alla migrazione delle cellule neoplastiche.
Pertanto, in coda alla chirurgia spesso è necessario un consolidamento degli esiti con la radioterapia e un controllo della malattia primitiva.